GFinisher. bioinformatics tool developed in the Graduate Program in Sciences - Biochemistry at the UFPR
GFinisher - Genome Finisher
Small manual
GFinisher is a finisher assembler tool that combines three tools:
- Misassemblies detection - points of maximum and minimum in Fuzzy GC Skew curve are used to identify probable spurious assemblies.
- jContigSort - ordering contig base and reference genome.
- jFGap - combine alternatives assemblies to close gap.
The application may be executed in automated or manually mode. In automatic mode
requires the availability of a reference genome.
- How to run complete pipeline - requires genome reference and two or more assemblies.
- How to run manual mode - run tools without genome reference.
How to run complete pipeline
For this small demonstration, to run the GFinisher is necessary to obtain assemblies and a reference genome. In this case study, we use the assemblies provided by GAGE-B genome of Bacteroides fragilis - HiSeq.
The download of assemblies can be made of the GAGE-B site or sourceforge. The reference genome can be download from NCBI or here.
If your computer doesn't have Java installed, try to download java and install it (www.java.com), preferably the 64-bit version.
Download the GFinisher binaries link
With all the resources available for you to run GFinisher, get to run.
Graphics mode
Run GFinisher in system console:
java -Xms2G -Xmx4G -jar GenomeFinisher.jar
At tab "Blast" verify the path where the blast was installed.
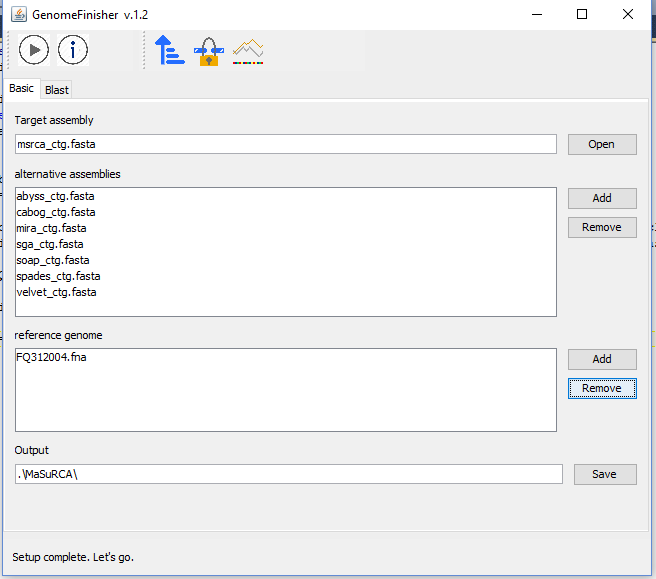
Go back to "Basic" tab and choose msrca_ctg.fasta file to "Target assembly" field and click at "Add" button in alternative assemblies section and choose files: abyss_ctg.fasta, cabog_ctg.fasta, mira_ctg.fasta, sga_ctg.fasta, soap_ctg.fasta, spades_ctg.fasta, velvet_ctg.fasta.
Add FQ312004.fna file in reference genome.
The figure below shows the data setup.
Click the first button in main toolbar and wait for about 15 minutes.
Console/Text mode
Run GFinisher in system console:
java -Xms2G -Xmx4G -jar GenomeFinisher.jar -config -create name_of_config.txt
Verify and adjust the settings on the name_of_config.txt, especially the path of the Blast and run
java -Xms2G -Xmx4G -jar GenomeFinisher.jar -config name_of_config.txt -i msrca_ctg.fasta \
-ref FQ312004.fna \
-ds abyss_ctg.fasta,cabog_ctg.fasta,mira_ctg.fasta,sga_ctg.fasta,soap_ctg.fasta,spades_ctg.fasta,velvet_ctg.fasta \
-o .\MaSuRCA\ -v 1
Results
When GFinisher is done all data generated stay at MaSuRCA folder. The table below show some reports produced by GFinisher in each steps. Click in image to expand view.
| Report type | Original data | After contigSort | After Broken FGC Skew | Results |
| N. contigs | 109 | 44 | 210 | 11 |
| N. contigs (≥ 500bp) | 109 | 44 | 210 | 11 |
| N. contigs (≥ 1000bp) | 102 | 44 | 210 | 11 |
| N50 | 158716 | 309306 | 72014 | 663436 |
| L50 | 11 | 6 | 23 | 3 |
| Dotplot |  |
 |
 |
 |
| GC Skew |  |
 |
 |
 |
| Quast | report | |||
Manual mode
Three tools may be executed individually:
- Misassemblies detection - points of maximum and minimum in Fuzzy GC Skew curve are used to identify probable spurious assemblies.
- jContigSort - ordering contig base and reference genome.
- jFGap - combine alternatives assemblies to close gap.
Misassemblies detection
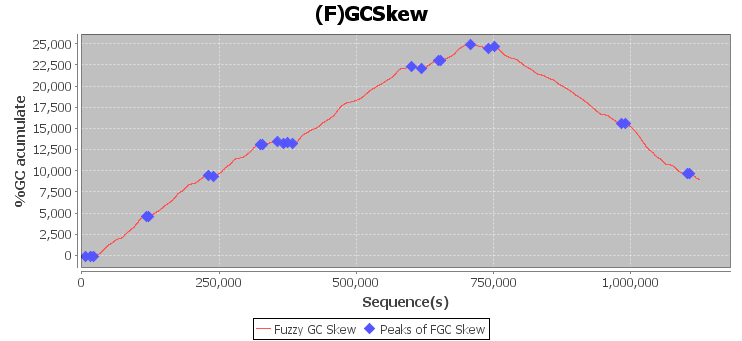
The Fuzzy GC Skew is used to identify possible errors in assembly contigs.
Follow the step by step how to perform the identification misassblies with GFinisher.
- Open GFinisher in graphics mode.
- click in Fuzzy GC Skew
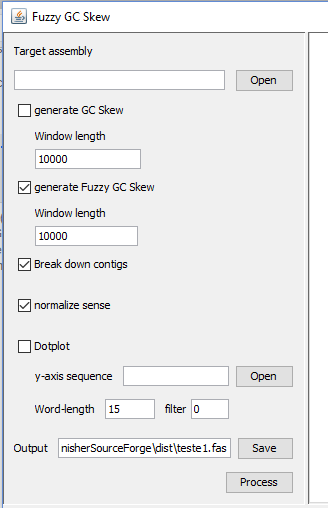
- Open your multi-fasta file (contigs/scaffolds)
- Select "generate Fuzzy GC Skew" and "Break down contigs" and "normalize sense" box
- The first run, we recommend 10,000bp for window length
- Choose the output filename
- Click in process button.
The option "normalize sense" is recommended when the assembler produces some small contigs length (less than window length parameter).
The GFinisher produces the result bellow.
jContigSort
The jContigSort is used to ordering contigs based on genome reference.
Follow the step by step how to.
- Open GFinisher in graphics mode.
- click in Contig Sort
- Open your multi-fasta file (contigs/scaffolds)
- Add genome reference
- Click in "run" button.
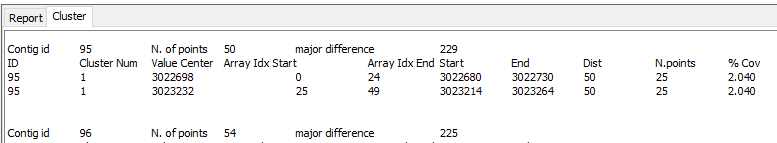
The GFinisher produces the result bellow. The first report shows the relative reference position of contigs and second report shows clusters of reference relative positions, this data can be to indicate duplicate region.

jFGap
jFGap is a tool that combine same assemblies to close gaps and improve assemblies,
originally version was written in Matlab (Gap).
- Open GFinisher in graphics mode.
- click in jFGap icon.
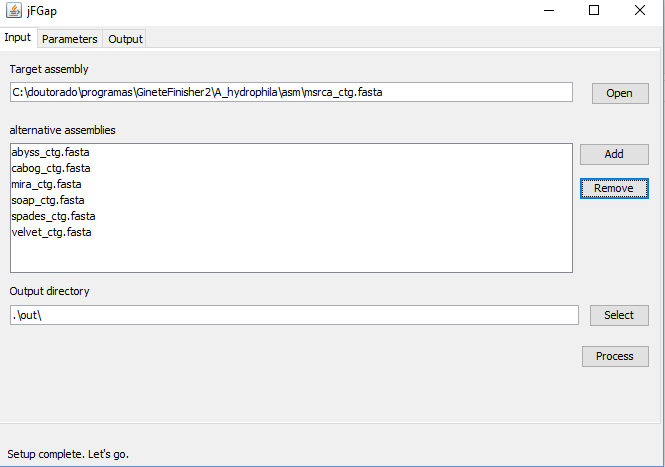
- Open your target assembly multi-fasta file (contigs/scaffolds)
- Add alternative assemblies
- Click in "process" button.
If "process button" are disabled, it's possible the paths setup is incomplete (see parameters tab).
The jFGap close only gaps identified by a specific symbol (see parameter tab) and jFGap don't change the order or sense of contigs, therefore recommends the use of jContigSort before running fgap.
